RED(RNA Editing Detector) is a program to detect and visualize RNA editing events at genomic scale using next-generation sequencing data. RED can also be used in command line. RNA editing is one of the post- or co-transcriptional processes with modification of RNA nucleotides from their genome-encoded sequence. In human, RNA editing event occurs mostly by deamination of adenosine to inosine (A-to-I) conversion through ADAR enzymes.
The main characteristics of RED are:
- Do Denovo or Non-Denovo(DNA-RNA) detection of RNA editing, while the former just uses RNA VCF file and the latter uses both RNA and DNA VCF file;
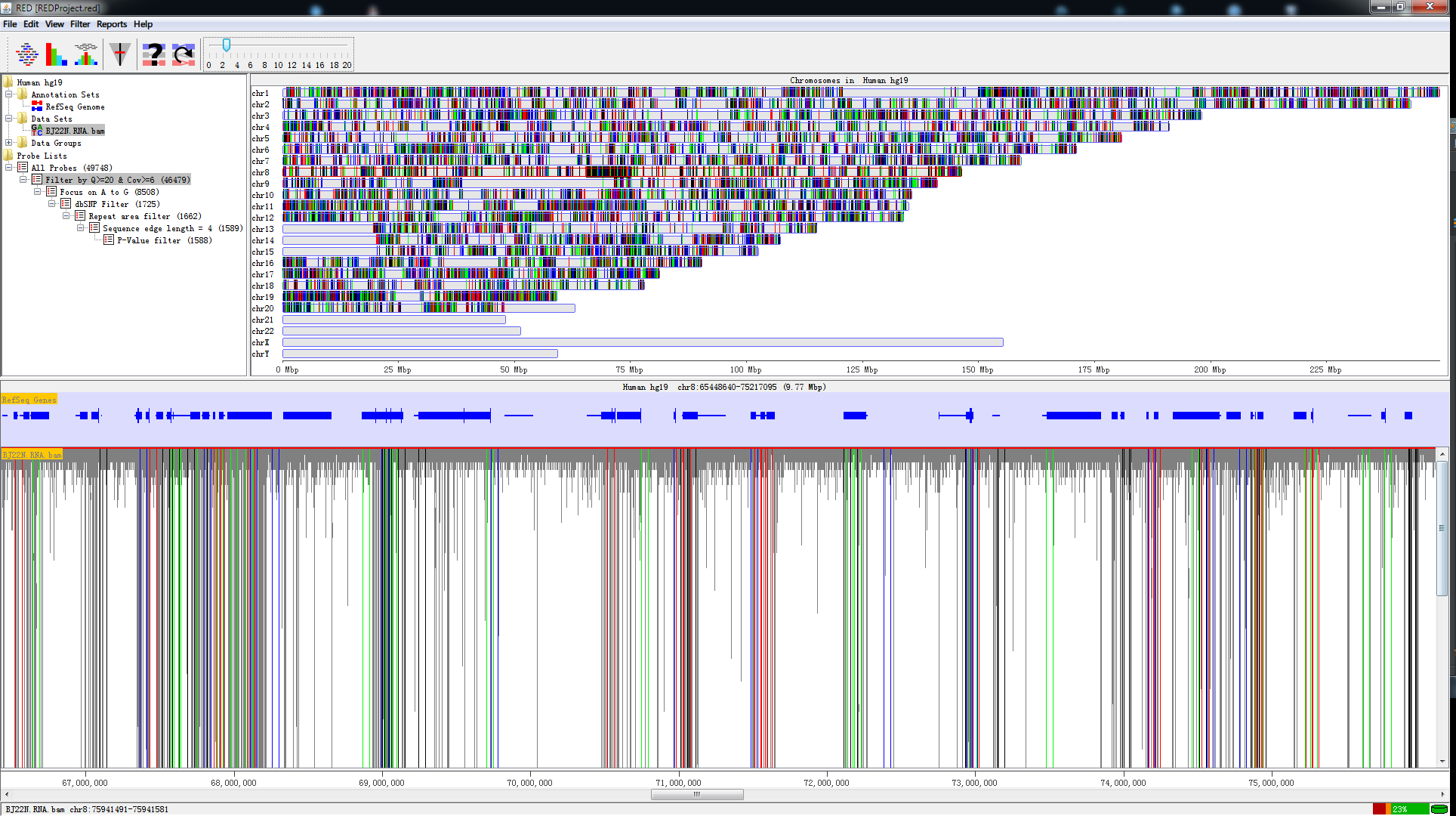
- Visualization of annotated genome with Reference Genes from UCSC and Reference Sequence from fasta file;
- Visualization of imported mapped raw data (BAM file) with genome scale and chromosome scale;
- Fast RNA editing detection speed with MySQL database;
- Visualization of editing sites with each step’s filter;
- Eight RNA editing site’s filters to filter out the most possible spurious sites.
- Quality Control Filter (contain quality filter and coverage filter);
- Editing Type Filter
- Splice Junction Filter;
- Known SNP Filter;
- Repeated Regions Filter;
- DNA-RNA Filter;
- Fisher's Exact Test Filter;
- Likelihood Ratio Test Filter.
To get started, check out http://redetector.github.io!
- Quick start
- Usage
- Documentation
- Contributing
- Bugs and feature requests
- Creators
- Copyright and license
Before letting RED work properly, the following software or program are required:
- Java Runtime Environment (tested on jdk 1.6.0_43 or later for Windows)
- MySQL Database Management System (tested on MySQL 5.6.19 or later for Windows)
- R Environment (tested on R 3.1.1 or later for Windows)
MySQL Database can be installed in a remote server, see Prerequisites for more detail.
Two quick start options for our software are available:
- Download the latest release.
- Download jar package.
- Clone the repo:
git clone https://github.com/REDetector/RED.git.
Within the download you'll find the following directories and files, logically grouping common assets. You'll see something like this:
./
├──lib
├──red
.gitignore
LICENSE.txt
README.md
Version.txt
Usage: java -jar RED.jarUsage: java -jar RED.jar [-h|--help] [-v|--version] [-H|--host[=127.0.0.1]] [-p|--port[=3306]] [-u|--user[=root]] [-P|--pwd[=root]] [-d|--database[=DNA_RNA_MODE]] [-m|--mode[=dnarna]] [-D|--delete] [-t|--type=AG] [-i|--input] [-o|--output[=./]] [-E|--export] [-O|--order[=123456[78]]] [--rnavcf] [--dnavcf] [--darned] [--splice] [--repeat] [--dbsnp]The most commonly used commands are:
-h, --help Print short help message and exit;
-v, --version Print version info and exit;
-H, --host=127.0.0.1 The host address of MySQL database;
-p, --port=3306 The port used in MySQL;
-u, --user=root MySQL user name;
-P, --pwd=root MySQL password of user;
-d, --database=DNA_RNA_MODE Database name, default is 'DNA_RNA_MODE';
-D, --delete=[sample] Delete the sample in database, delete all samples if [sample]=all;
-m, --mode=dnarna Tell the program if it is denovo mode or DNARNA mode [DENOVO|DNARNA];
-E, --export=[data] Export final result for a given database, [data] can be
all | annotation | any column of the final table in database,
divided by ',', such as [data]=chrom,pos,ref,alt,pvalue;
-t, --type=AG Substitution type that focus on, such as 'AG','CT','GC' etc, all
substitution types will be calculated if [type]=all
-r, --r=/usr/bin/RScript File path of RScript or RExecutable.
-i, --input Input all required files (i.e., RNA VCF File, DNA VCF File, DARNED Database,
Gene Annotation File, RepeatMasker Database File, dbSNP Database File, RADAR
Database) instead of single input, each file should be divided with ',', and
start with reference file name, for example, 'rnavcf-/data/RED/BJ22.snvs.vcf';
-o, --output= Set export path for the results in database, default path is current
directory;
-e, --export=all Export the needed columns in database, which must be the column name
of a table in database, the column names should be divided by ',';
-O, --order=12345678 The order of performing the filter.
--rnavcf File path of RNA VCF file;
--dnavcf File path of DNA VCF file;
--darned File path of DARNED database;
--splice File path of annotation genes like "gene.gft";
--repeat File path of Repeat Masker database;
--dbsnp File path of dbSNP database;
###Explanation for order option
The default order for DNA-RNA mode is (1)editing type filter -> (2)quality control filter -> (3)DNA-RNA filter -> (4)splice junction filter -> (5)repeat regions filter -> (6)known SNP filter -> (7)likelihood ratio test filter -> (8)fisher's exact test filter.
The default order for denovo mode is (1)editing type filter -> (2)quality control filter -> (3)splice junction filter -> (4)repeat regions filter -> (5)known SNP filter -> (6)fisher's exact test filter.
You can change the order by this option. For DNA-RNA mode, eight filters are available so that you could not enter less than 8 in the order (e.g., '2143657' is illegal, '51432678' is legal). The same for denovo mode, six filters are available (e.g., '5214376' is illegal, '523516' is legal).
It is strongly recommended the FET filter place in the last of the order since it will affect the results by calculating the p-value and false discovery rate.
Besides, if there is any filter that you do not want to perform in the filter list, just replace the index number from the filter name to zero. For example, in DNA-RNA mode, I do not want to perform known SNP filter and likelihood test filter, then the order should be '12345008'. You can change the filter order, too.
###Example:
-
- In Windows, use '--' patterns, focus on A-to-G editing type, perform denovo mode and export all data.
java -jar E:\Workspace\RED\out\artifacts\RED\RED.jar ^
--host=127.0.0.1 ^
--port=3306 ^
--user=root ^
--pwd=123456 ^
--mode=denovo ^
--type=AG ^
--input=rnavcf-D:\Downloads\Documents\BJ22.snvs.hard.filtered.vcf,darned-D:\Downloads\Documents\hg19.txt,splice-D:\Downloads\Documents\genes.gtf,repeat-D:\Downloads\Documents\hg19.fa.out,dbsnp-D:\Downloads\Documents\dbsnp_138.hg19.vcf,radar-D:\Downloads\Documents\Human_AG_all_hg19_v2.txt ^
--output=E:\Workspace\RED\Results ^
--export=all ^
--r=C:\R\R-3.1.1\bin\Rscript.exe-
- In Windows, use '-' patterns, focus on all editing types, perform dnarna mode, change the filter order, then export chromosome, position and editing level.
java -jar E:\Workspace\RED\out\artifacts\RED\RED.jar ^
-H 127.0.0.1 ^
-p 3306 ^
-u root ^
-P 123456 ^
-d BJ22_DNA_RNA ^
-m dnarna ^
-O 12354678 ^
-i rnavcf-D:\Downloads\Documents\BJ22.snvs.hard.filtered.vcf,dnavcf-D:\Downloads\Documents\BJ22_sites.hard.filtered.vcf,darned-D:\Downloads\Documents\hg19.txt,splice-D:\Downloads\Documents\genes.gtf,repeat-D:\Downloads\Documents\hg19.fa.out,dbsnp-D:\Downloads\Documents\dbsnp_138.hg19.vcf,radar-D:\Downloads\Documents\Human_AG_all_hg19_v2.txt ^
-o E:\Workspace\RED\Results ^
-E chrom,pos,level ^
-r C:\R\R-3.1.1\bin\Rscript.exe-
- In CentOS, use hybrid '-' and '--' patterns, focus on C-to-T editing type, perform denovo mode, and export nothing (only stored in database).
java -jar /home/seq/softWare/RED/RED.jar
-h 127.0.0.1 \
-p 3306 \
-u seq \
-P 123456 \
-d BJ22_DENOVO \
-m denovo \
-t CT \
--rnavcf=/data/rnaEditing/GM12878/GM12878.snvs.hard.filtered.vcf \
--repeat=/home/seq/softWare/RED/hg19.fa.out \
--splice=/home/seq/softWare/RED/genes.gtf \
--dbsnp=/home/seq/softWare/RED/dbsnp_138.hg19.vcf \
--darned=/home/seq/softWare/RED/hg19.txt \
--radar=/home/seq/softWare/RED/Human_AG_all_hg19_v2.txt \
--r=/usr/bin/Rscript-
- In CentOS, delete sample 'BJ22' in database;
java -jar /home/seq/softWare/RED/RED.jar
-h 127.0.0.1 \
-p 3306 \
-u seq \
-P 123456 \
-m denovo \
-d BJ22_DENOVO \
-D BJ22File->New->Project from Existing Sources...
- Select File or Directory to Import
- Create project from existing sources
- Enter Project Name
- Mark All, Next
- Tick
liblibraries, Next - Tick
REDmodueles, Next - Select Project SDK (Default JDK: jdk1.6.0_43)
- Finish
File->New->Java Project
- Enter Project Name: RED
- Untick the
Use default locationand set location to this project, Next - Finish.
Test data for RED is available here.
RED's documentation, included in this repo in the root directory, is built with Jekyll and publicly hosted on GitHub Pages at http://redetector.github.io. The docs can also be run locally.
The documentation contains following sections:
- Introduction
- Getting Started
- Visualization
- Filters
- Reports
- Saving Data
- Configuration
- Reporting Bugs
- Credits
Please move to pages at http://redetector.github.io for more information.
Previous releases and their documentation are also available for download.
Please read through our contributing guidelines.
Have a bug or a feature request? Please first read the issue guidelines and search for existing and closed issues . If your problem or idea is not addressed yet, please open a new issue.
Xing Li
- https://github.com/iluhcm
- email:
echo "c2FtLmx4aW5nQGdtYWlsLmNvbQ==" | base64 -D
Di Wu
Code and documentation copyright 2013-2014 BUPT/CQMU.
RED is a free software, and you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 3 of the License, or (at your option) any later version .
SeqMonk & Integrative Genome Viewer (IGV)
We thank SeqMonk and IGV. The framework of the GUI in RED is based on SeqMonk, whose GUI is very brief and operating efficiency is fairly high. Meantime, the genome annotation data (mainly referred to gene.txt/gene.gtf) in Feature Track is obtained from genome server of IGV when there is no genome file in the local host.